![]()



یک فرصت پسادکتری در دانشگاه شهیدرجایی زیر نظر فدراسیون سرآمدان ایران در حال حاضر باز می باشد که علاقه مندان می توانند با مکاتبه با ایمیل زیر جهت استفاده از این فرصت اعلام آمدگی نمایند.
This email address is being protected from spambots. You need JavaScript enabled to view it.
این فرصت زیر نظر دکتری بهشتیان از گروه شیمی دانشگاه شهیدرجایی مدیریت خواهد شد. داوطلبان این فرصت پسادکتری می بایست فارغ التحصیل رشته ی شیمی محاسباتی، فیزیک محاسباتی، برق، متالوژی و یا رشته های مربوطه باشد. داوطلب باید حتما تجربه ی قوی ای در زمینه ی علوم محاسباتی و کار با نرم افزار های محاسباتی ای مانند، Quantum ESPRESSO، SIESTA, VASP, Material Studio و .... داشته باشد و با مباحث اولیه ی مربوط به تئوری تابعی چگالی آشنایی کامل داشته باشد. داشتن مقالات سطح بالا در این حوزه بسیار ضروری است و همچنین تسلط قوی به زبان انگلیسی ضروری می باشد.
حوزه های مطالعاتی گروه شبیه سازی دکتری بهشتیان در زمینه های زیر می باشد:
- باتری های لیتیوم/سدیم -یون
- ابرخازن ها
-ابررساناها
-فیلتر ها
می باشد. لذا داوطلب این فرصت پسادکتری، می بایست آشنایی اولیه با این مباحث داشته باشد.
از انجایی که امکانات خوابگاهی در اختیار نیست داوطلب می بایست ساکن تهران باشد. علاوه بر این با توجه به قوانین فدراسیون سرآمدان، این فرصت در اختیار فردی قرار می گیرد که شغل دیگری نداشته و از جای دیگری برای وی بیمه پرداخت نمی شود. همچنین، این فرصت، تمام وقت بوده و محقق می بایست 5 روز هفته از ساعت 9 الی 17 در محل کار در دانشگاه شهید رجایی حضور داشته باشد.
دانشگاه تربیت دبیر شهید رجایی دانشگاهی دولتی در شهر تهران است که وابسته به وزارت آموزش و پرورش و تحت نظر وزارت علوم تحقیقات و فناوری میباشد. این دانشگاه با مساحتی نزدیک به ۱۵۰٬۰۰۰ مترمربع در منطقه لویزان واقع در شمال شرق تهران قرار دارد و مجموعاً دارای بیش از ۵۶٬۰۰۰ متر مربع فضای سرپوشیدهٔ آموزشی، آزمایشگاهی، کارگاهی، رفاهی و فرهنگی است. در گذشته وظیفه اصلی این دانشگاه تربیت دبیر فنی و نیروی انسانی مورد نیاز آموزش و پرورش بود ولی در حال حاضر علاوه بر تربیت دبیر فنی مانند سایر دانشگاه های دولتی به تربیت دانشجو در مقاطع مختلف تحصیلی مشغول است. روش پذیرش دانشجو نیز همانند همه دانشگاههای دولتی از طریق آزمون سراسری توسط سازمان سنجش و آموزش کشور میباشد.


CARBON یک توزیع آماده گنو/لینوکسی است که در چندین نسخه، از بستههای نرمافزاری مختلف در زمینهی علم مواد پشتیبانی میکند.
هدف از تولید CARBON این بوده است که شما بدون نیاز به طی کردن فرآیند پیچیدهی نصب و آمادهسازی نرمافزارهای محاسباتی، بتوانید روی رایانه خود به طور آماده با آنها کار کرده و تمام تمرکز خود را روی شبیهسازی و محاسباتتان بگذارید.
کاری که تیم توسعهی نرمافزارهای محاسباتی در NRTC انجام داده این بوده که یک توزیع گنو/لینوکسی ساخته که پکیج محاسباتی مورد نظر به صورت بهینه روی آن نصب شده است. این توزیع محاسباتی قابلیت این را دارد که به روی سیستمعامل شما (چه با GNU/Linux کار کنید و چه کاربر Windows باشید) به صورت یک ماشین مجازی اجرا شود، بدون اینکه با سیستم اصلی شما تداخل داشته باشد.
در این راهنما به طور کامل با راهاندازی CARBON روی رایانه خود آشنا میشوید.
راهنمای کاربران GNU/Linux
برای استفاده از CARBON شما باید دو برنامهی virtualbox و vagrant را دریافت کنید تا بتوانید بواسطه آنها توزیع محاسباتی CARBON را به طور مجازی روی سیستم خود اجرا و مدیریت کنید.
دقت کنید که هردوی این سایتها تحریم هستند و شما برای دانلود برنامهها نیاز به vpn دارید.
ابتدا آخرین نسخه virtualbox را مطابق با سیستم عامل خود از آدرس زیر دریافت کنید:
https://www.virtualbox.org/wiki/Downloads
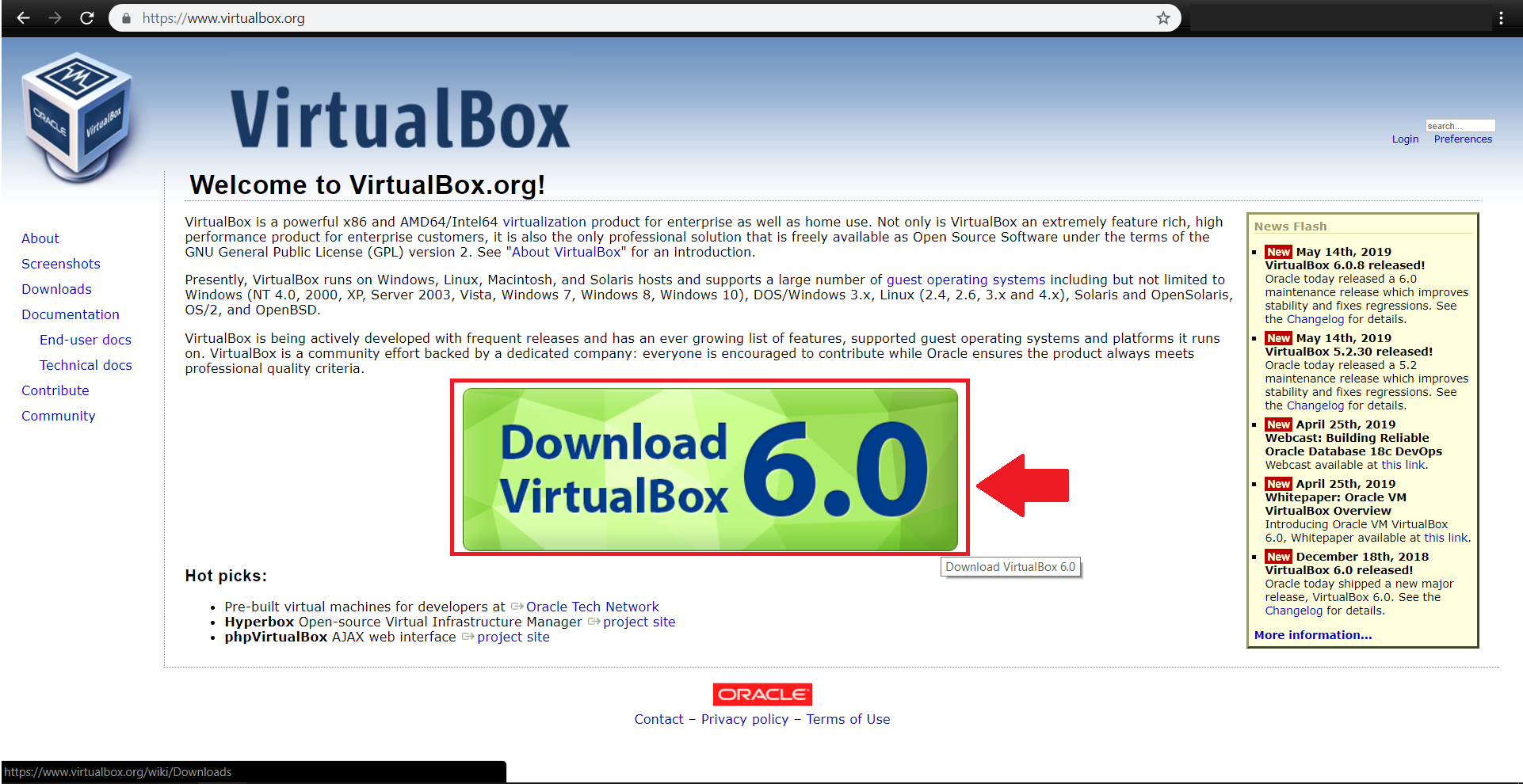
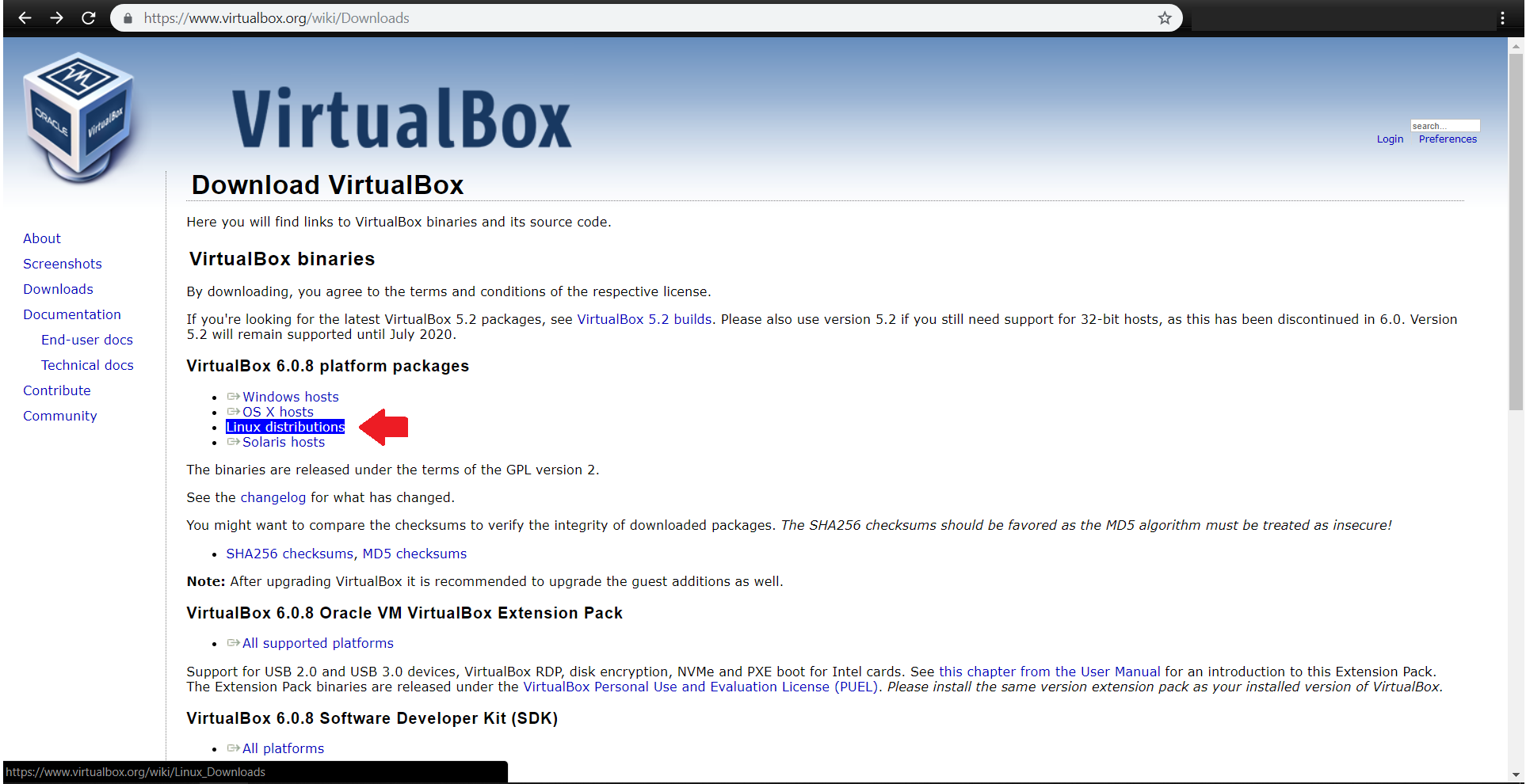
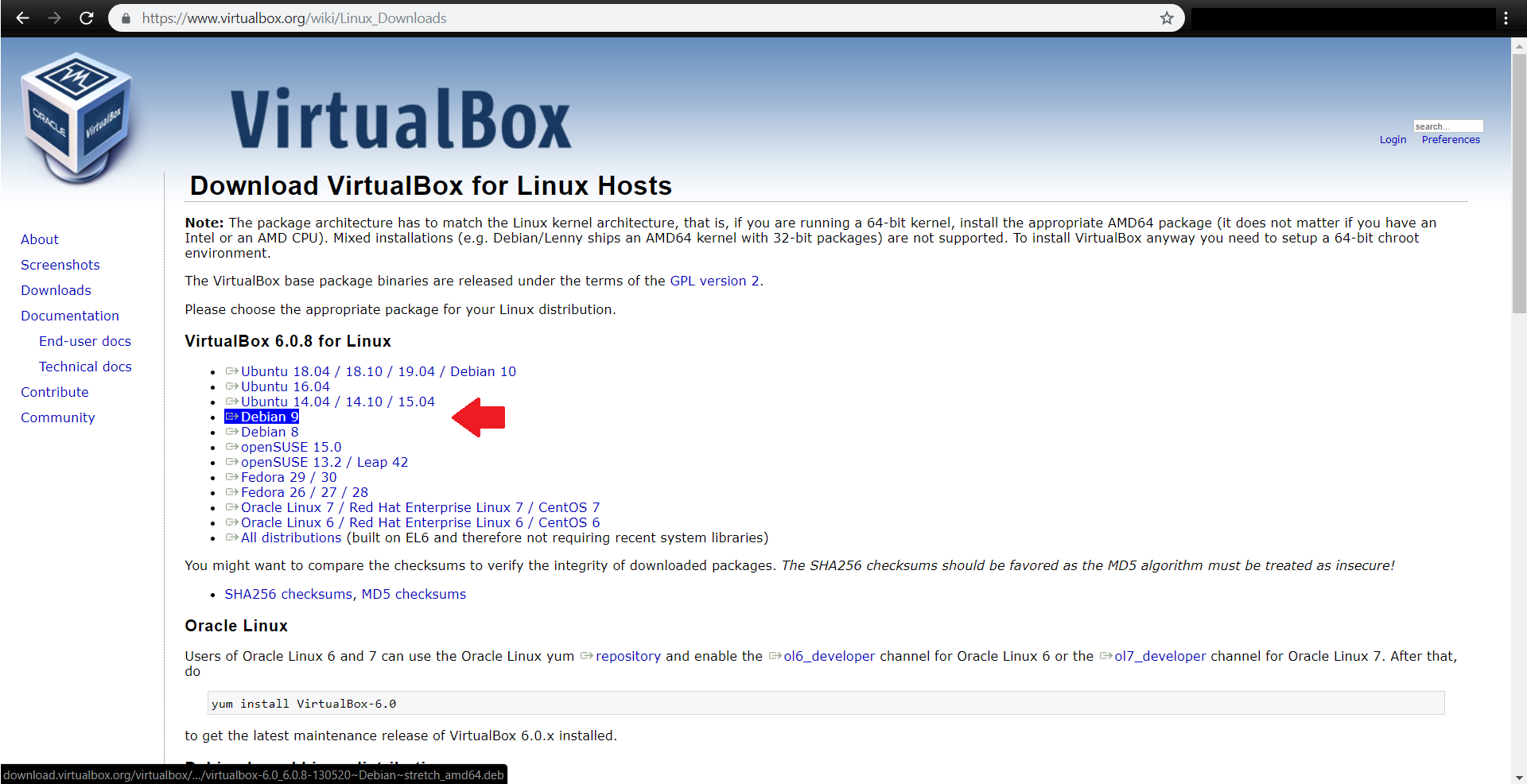
فایل virtualbox-6.0_6.0.8-130520_Debian_stretch_amd64.deb دانلود شده و در مسیر پیشفرض Download قرار میگیرد.
با دستورات زیر در مسیر جاری برنامه قرار گرفته و برنامه را نصب میکنیم:
$ cd Download
![]()
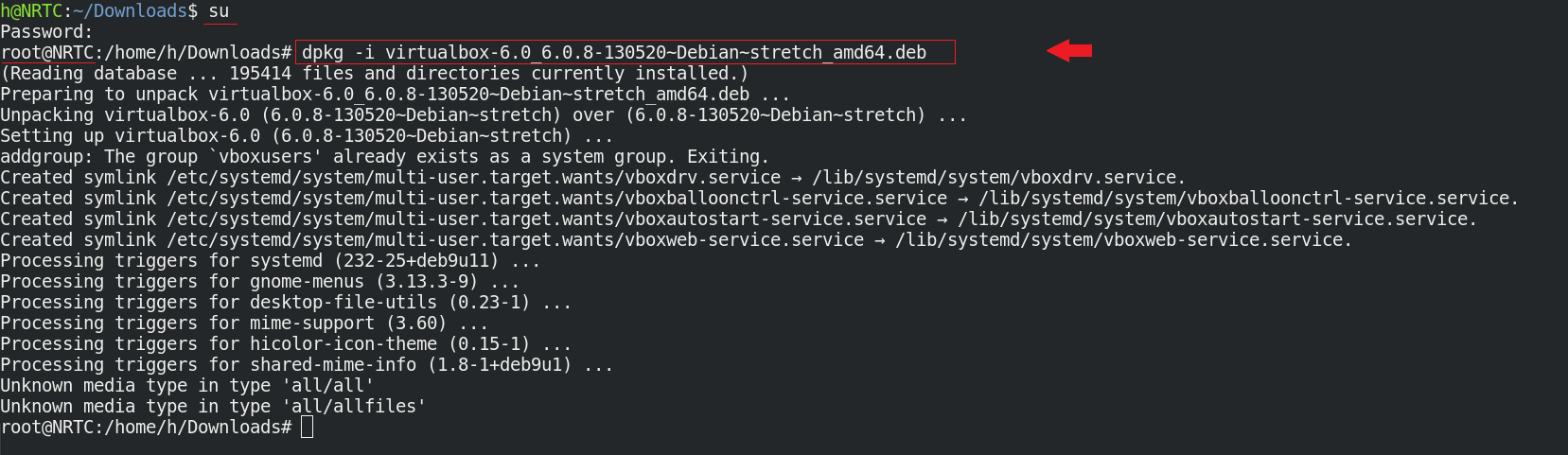
دقت کنید که برای نصب، نیاز به دسترسی کابر ریشه (root) دارید:
$ sudo dpkg -i virtualbox-6.0_6.0.8-130520~Debian~stretch_amd64.deb

و یا
همچنین vagrant را نیز مطابق با سیستم عامل خود از آدرس زیر دریافت و نصب کنید:
https://www.vagrantup.com/downloads.html
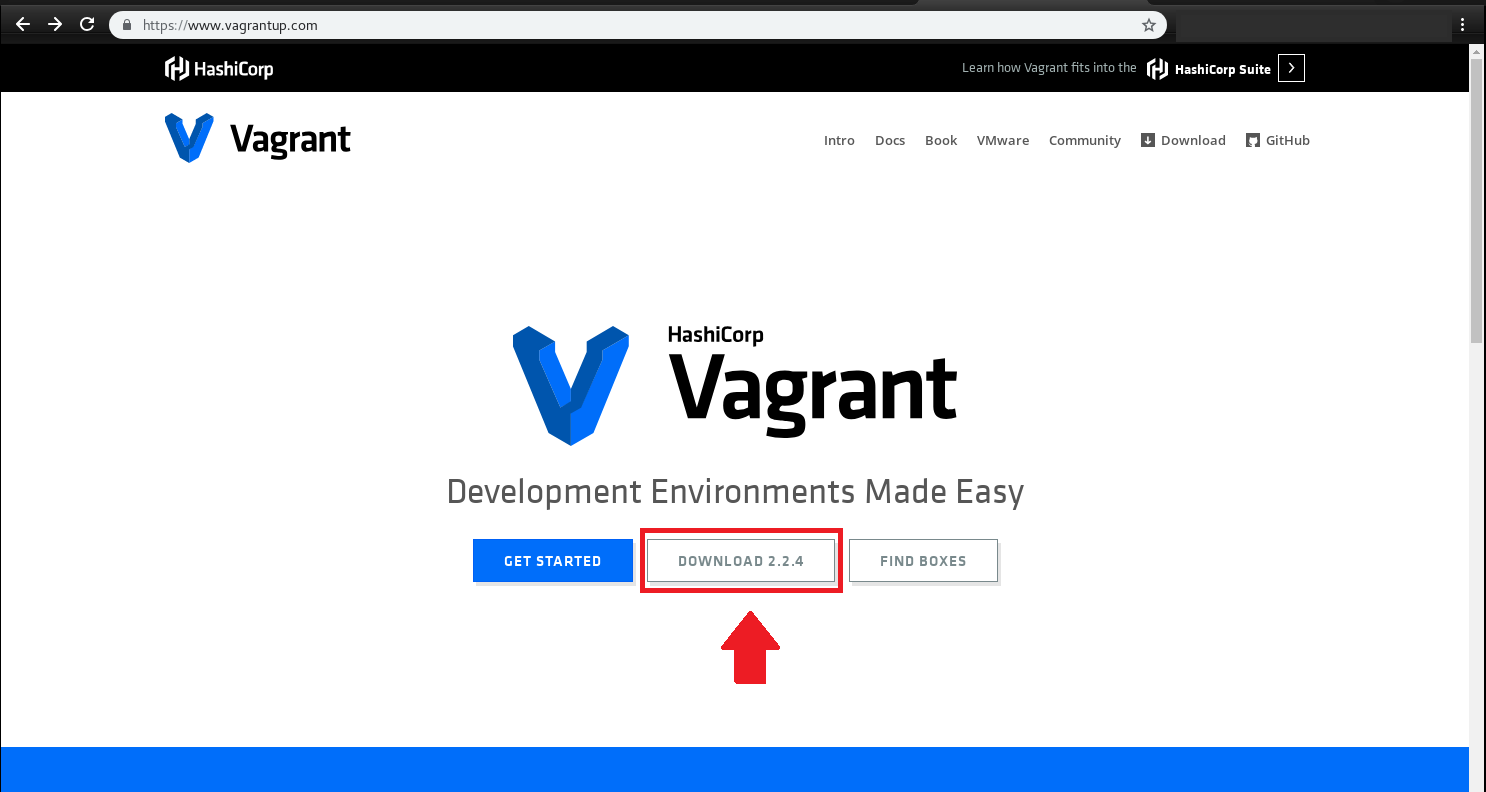
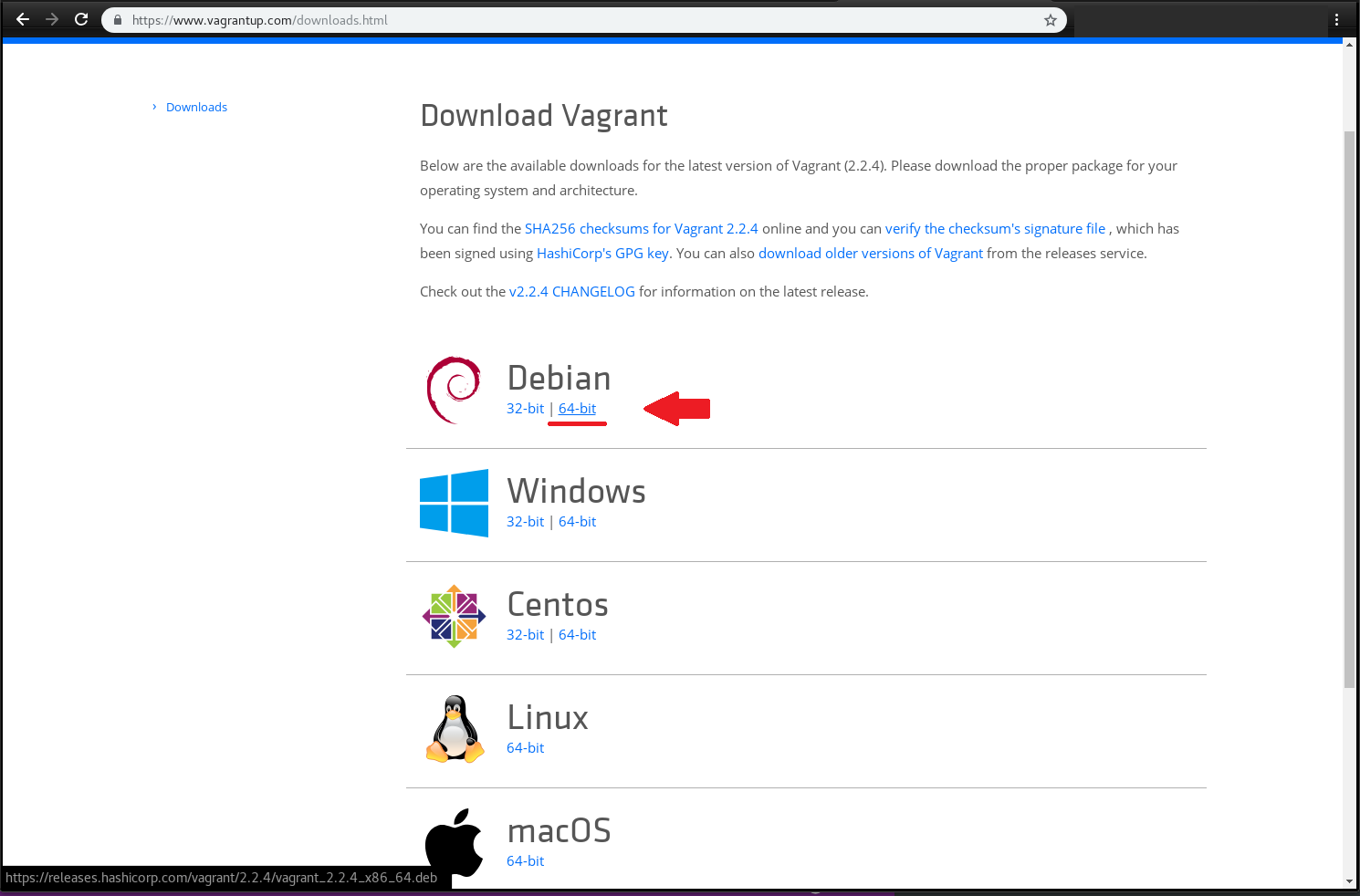
$ cd /Download
$ dpkg -i vagrant_2.2.4_x86_64.deb
![]()
![]()
و یا
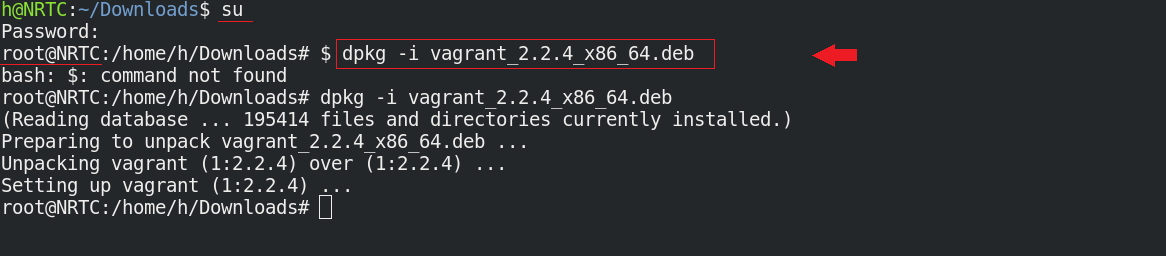
حال به پوشهای که دیتاهای محاسباتی شما در آن است بروید و دستور زیر را وارد کنید:
$ vagrant init nrtc/carbon-qe-intel --box-version 0.2
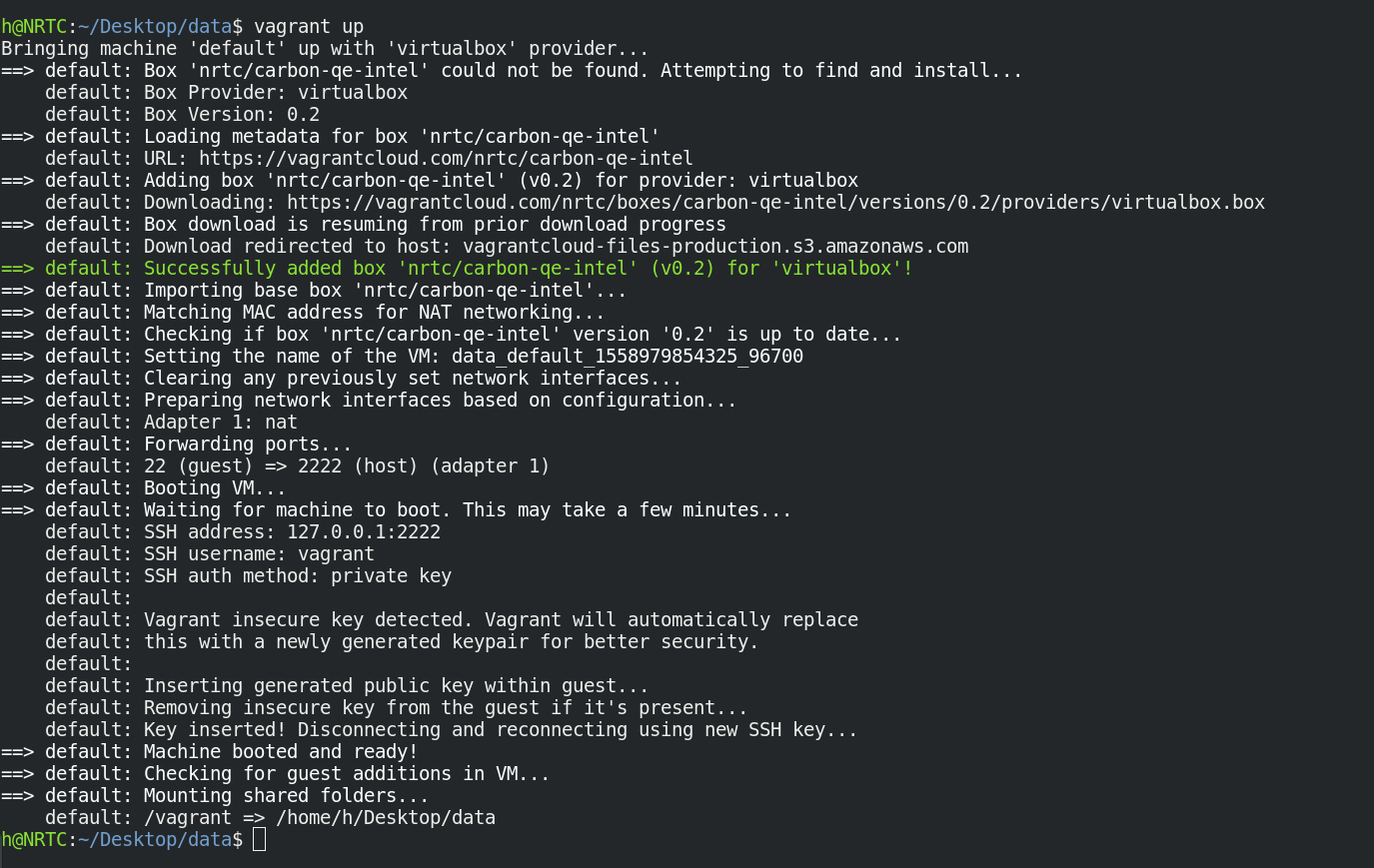
$ vagrant up
![]()
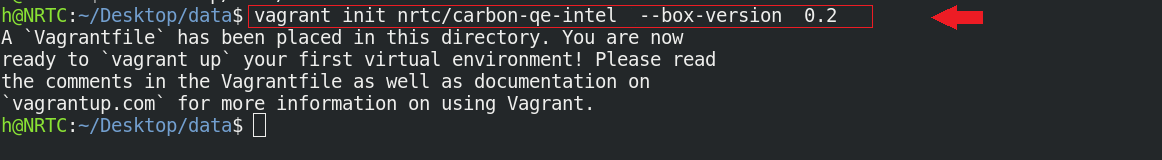
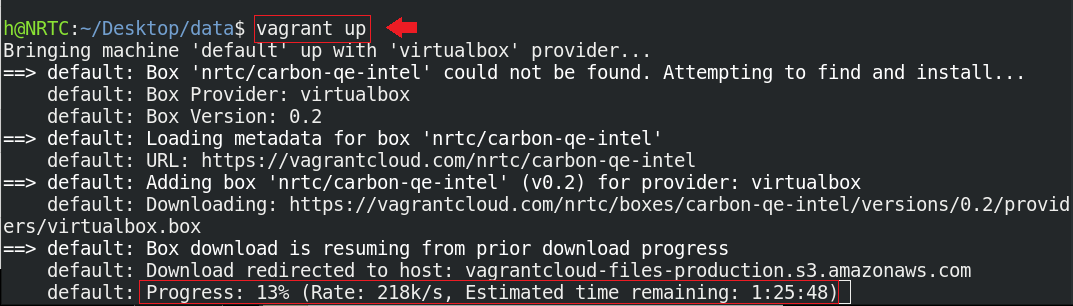
در این مرحله باید اندکی صبر کنید تا باکس کربن از vagrant cloud دریافت شود. بهتر است که حوصلهی شما متناسب با معکوس سرعت اینترنتتان باشد!
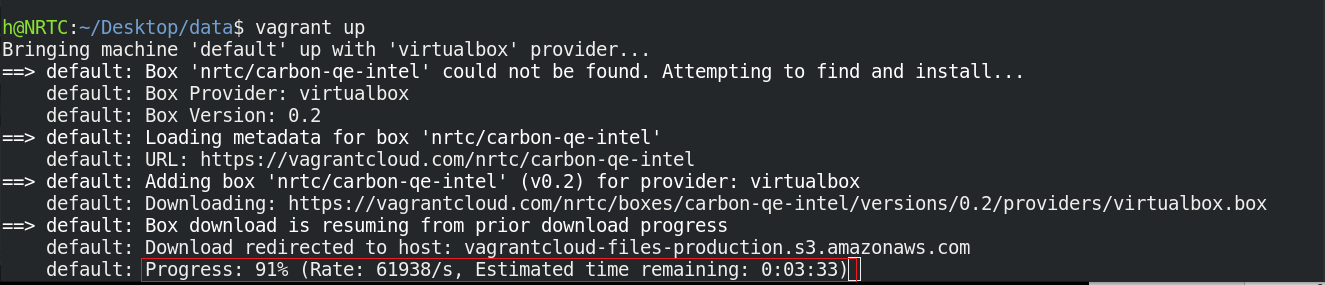
اکنون توزیع محاسباتی CARBON بر روی رایانه شما راه اندازی شده است.
فقط کافی است تا با دستور زیر وارد محیط CARBON شوید و مدیریت ماشین محاسباتی خود را در دست بگیرید:
$ vagrant ssh
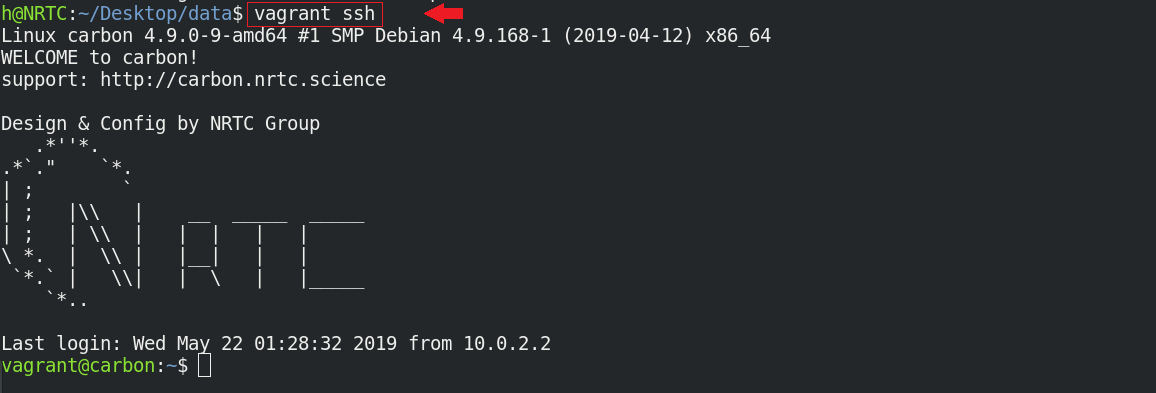
میبینید که خطفرمان شما از محیط سیستم عامل اصلی، به محیط سیستمعامل محاسباتی CARBON تغییر کرده است.
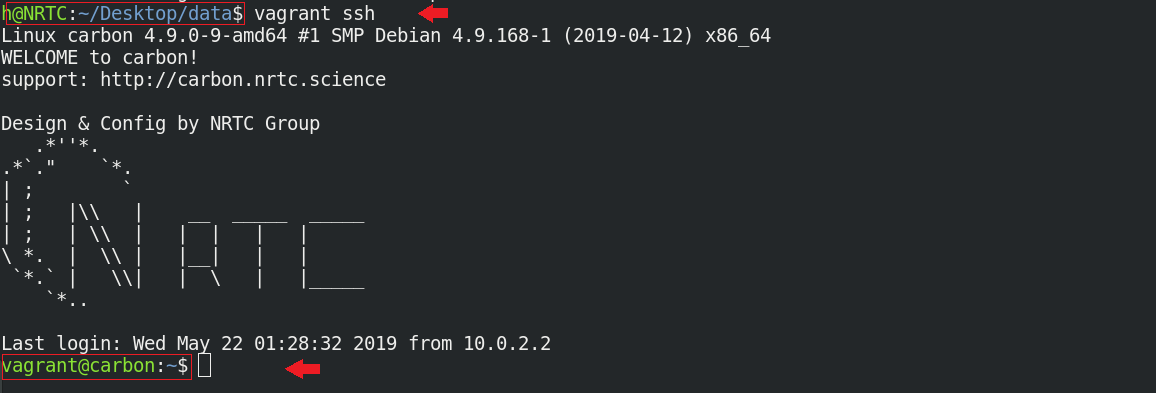
شما هم اکنون میتوانید در محیط جدید، بدون نیاز به طی کردن مسیر پردردسر نصب برنامه، از نرم افزار محاسباتی مد نظر به سادگی استفاده کنید.
برای اینکه به دادههای خروجی و نتایج محاسباتتان در سیستم عامل اصلی دسترسی داشته باشید، کافیست تمام اطلاعاتتان را در ماشین مجازی در دایرکتوری /vagrant ذخیره کنید تا در همان مسیر /data در سیستم عامل اصلی آنها را بیابید.
vagrant@nrtc/carbon:$ /vagrant

برای خروج از CARBON و دسترسی به خطفرمان سیستم اصلی خود،کافی است که exit را در ترمینال تایپ کنید:
$ exit

ویا از کلید میانبر "ctrl+D" استفاده کنید.
پس از خروج از ماشین مجازی CARBON، برای خاموش کردن ماشین، از
$ vagrant halt
استفاده کنید.

دادههای محاسباتی و همه تغییرات شما در سیستم عامل مجازی CARBON ذخیره شده است.
برای ورود و خروجهای بعدی کافی است دستورات زیر را پیش بگیرید:
$ vagrant up
$ vagrant ssh
انجام محاسبات
$ exit
$ vagrant halt
راهنمای کاربران Windows
برای استفاده از CARBON شما باید دو برنامهی virtualbox و vagrant را دریافت کنید تا بتوانید بواسطه آنها توزیع محاسباتی CARBON را به طور مجازی روی سیستم خود اجرا و مدیریت کنید.
همچنین باید با محیط powershell در ویندوز خود کار کنید.
ابتدا آخرین نسخه virtualbox را مطابق با سیستم عامل خود از آدرس زیر دریافت کنید:
https://www.virtualbox.org/wiki/Downloads
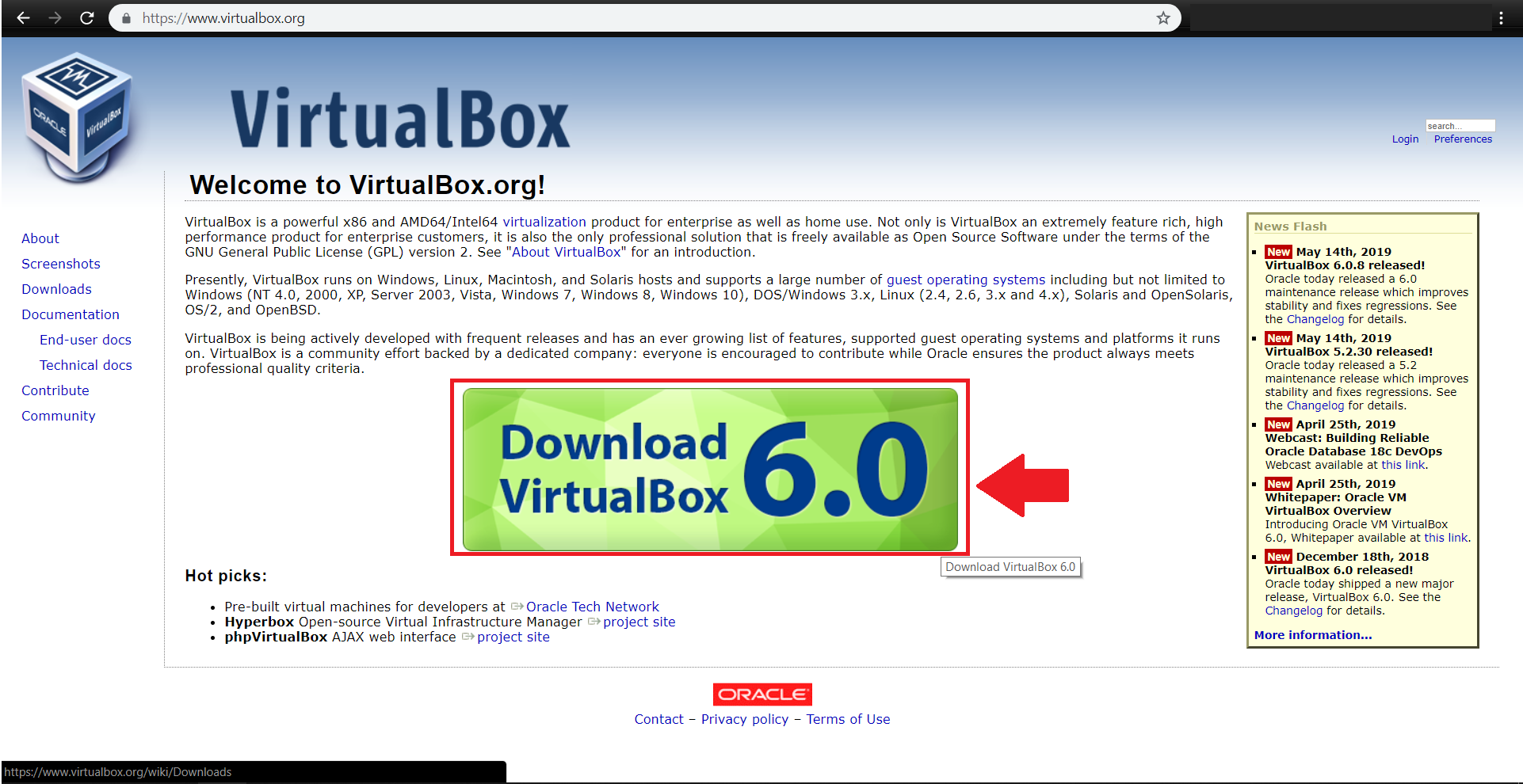
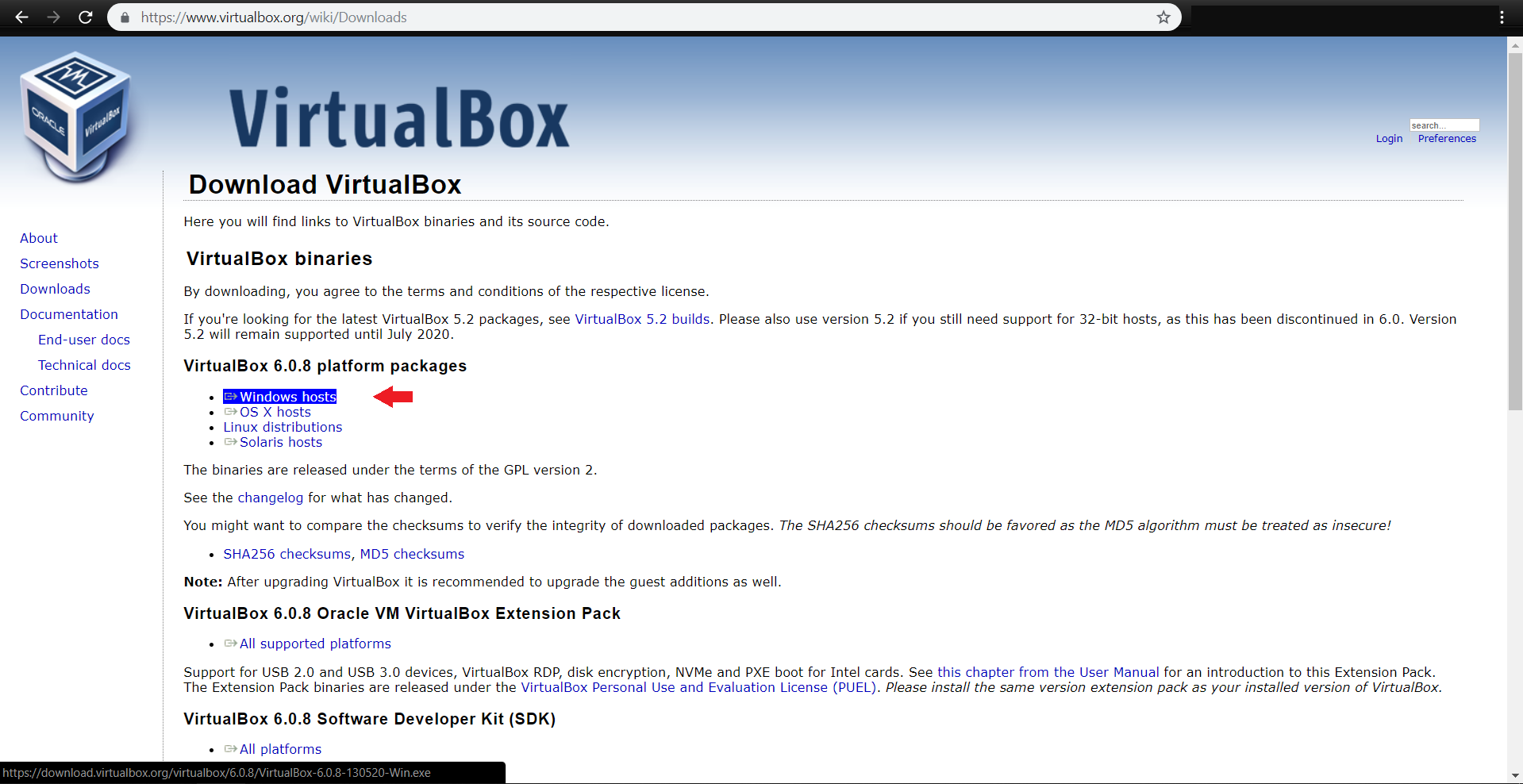
همچنین vagrant را نیز مطابق با سیستم عامل خود از آدرس زیر دریافت نمایید:
https://www.vagrantup.com/downloads.html
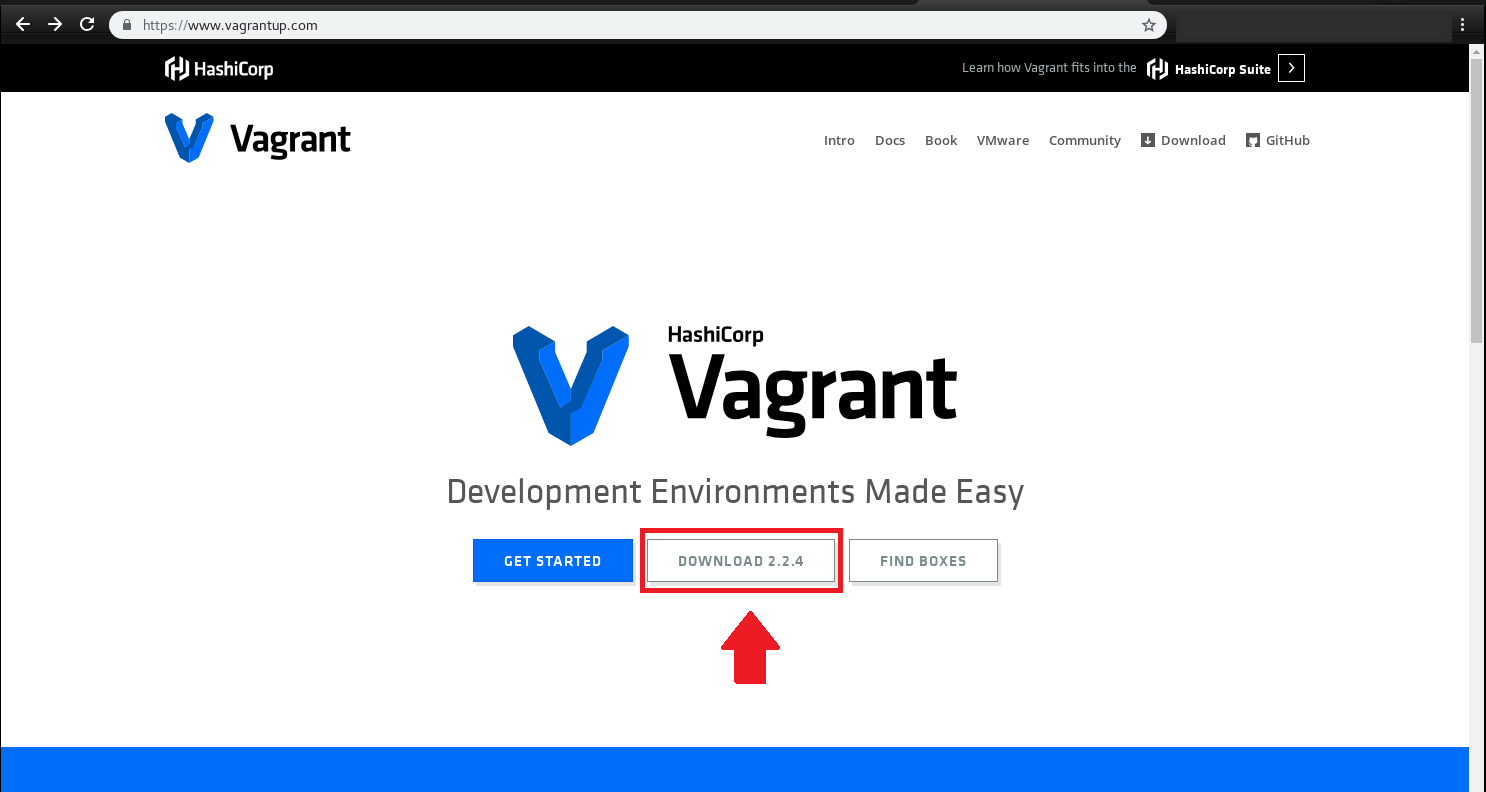
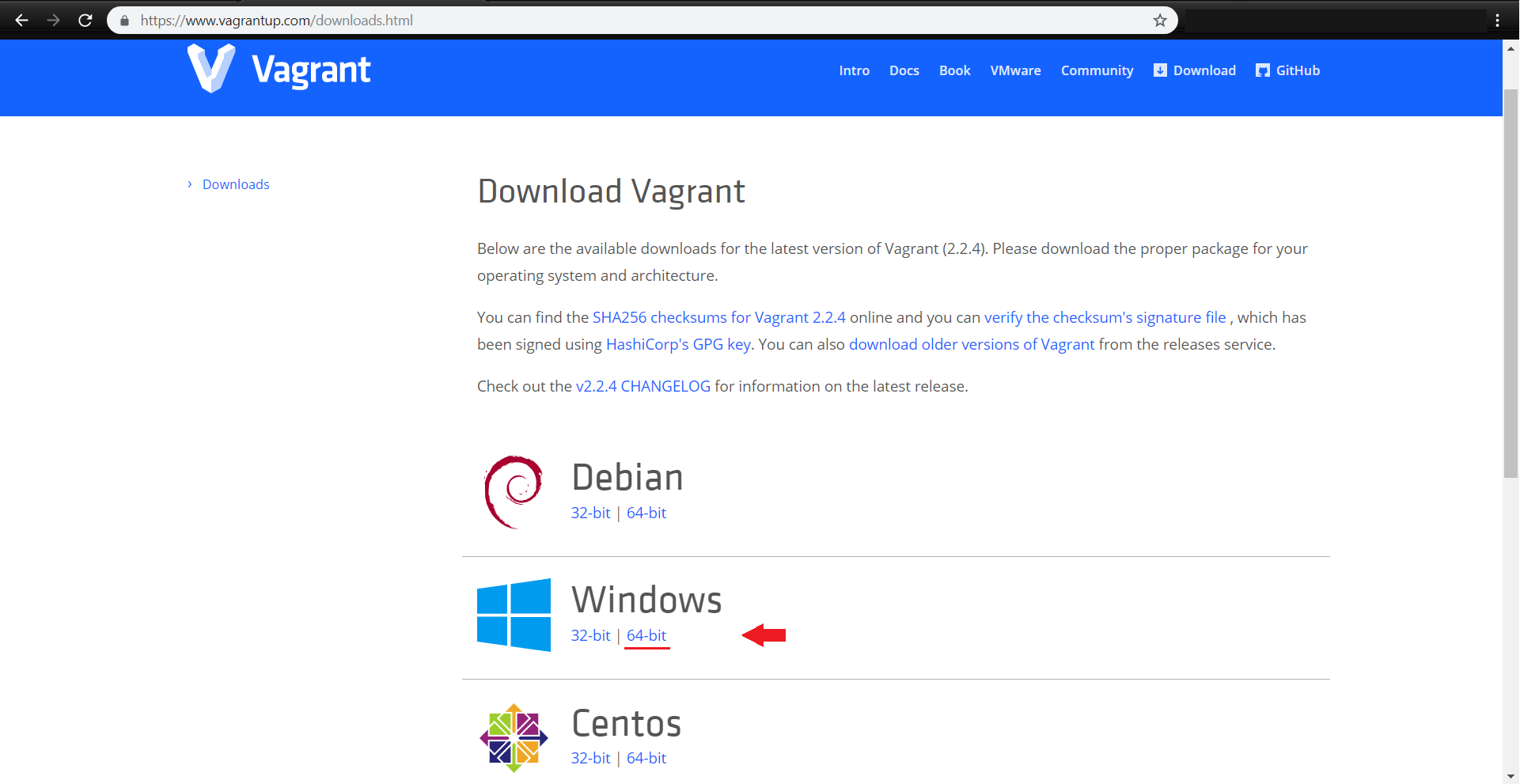
برنامههای دانلود شده را طبق روال نصب کنید:
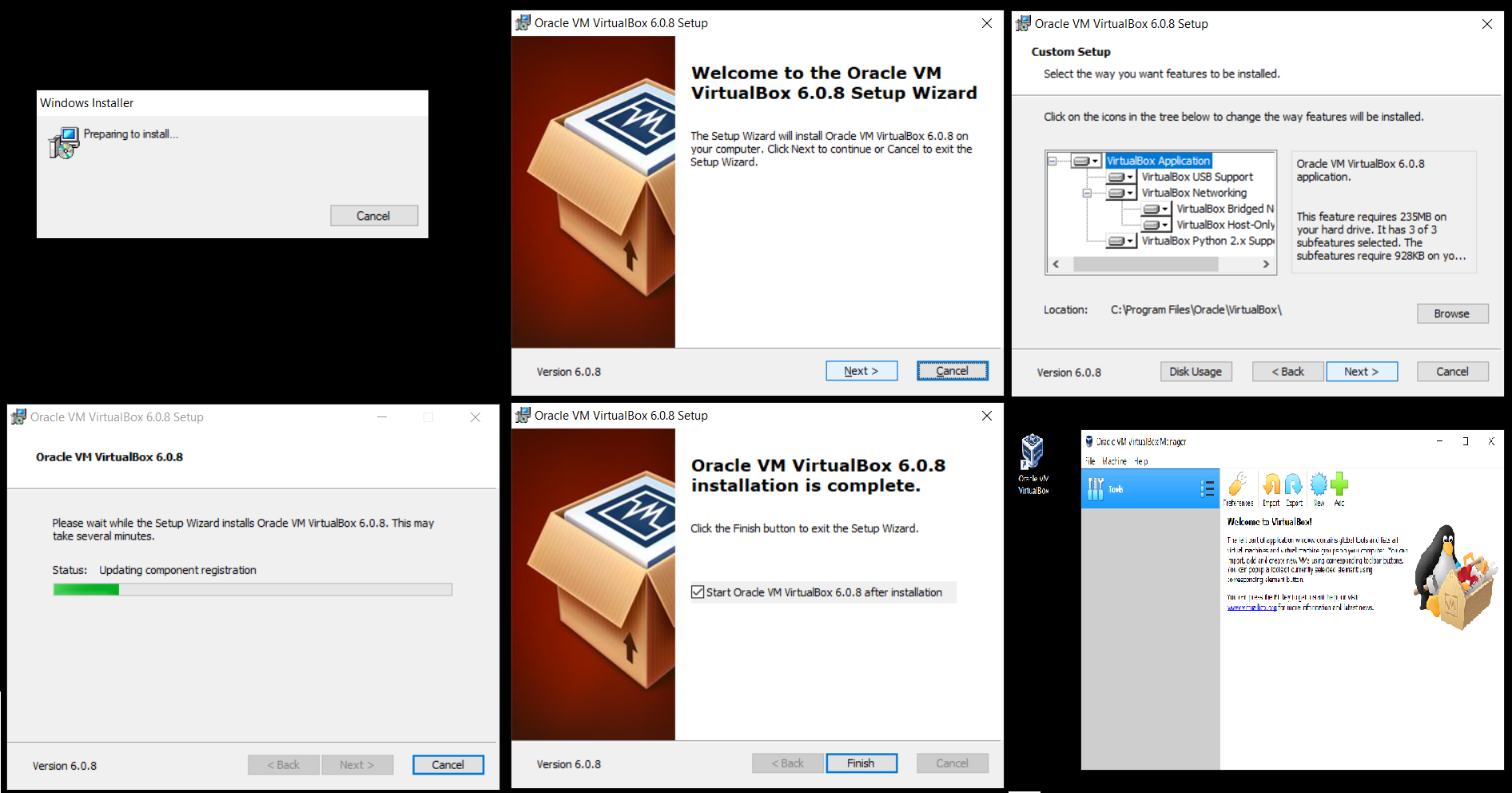
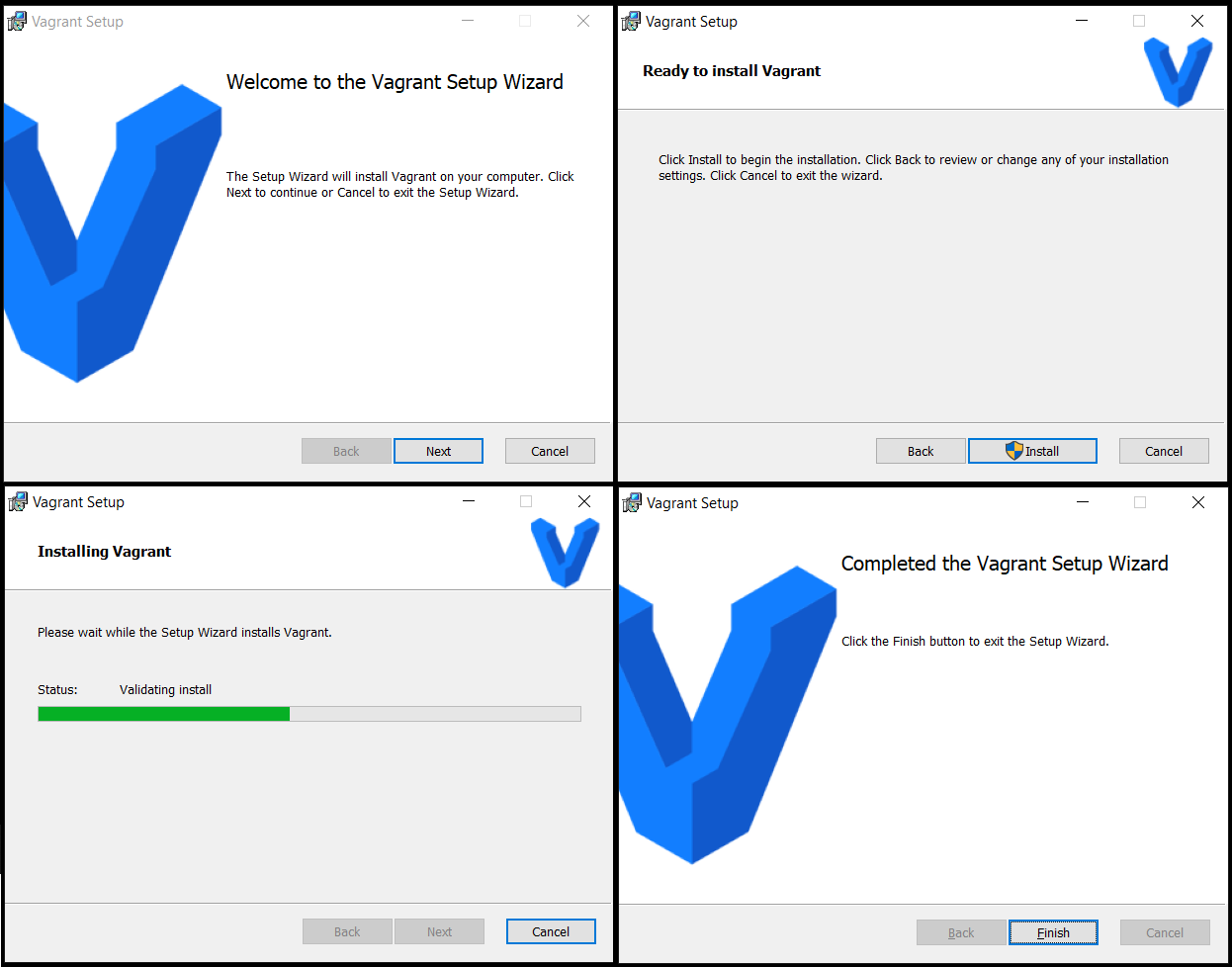
پس از اتمام مراحل نصب، powershell ویندوز را باز کنید و به پوشهای که دیتاهای محاسباتی شما در آن است بروید،
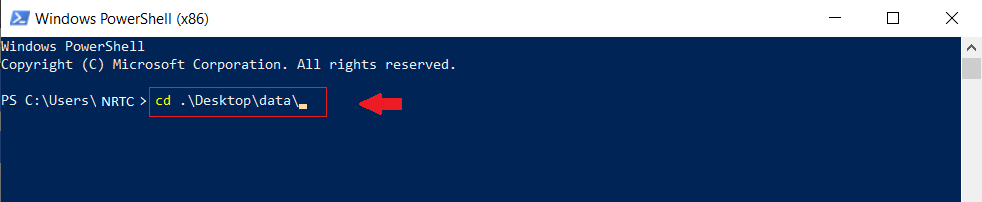
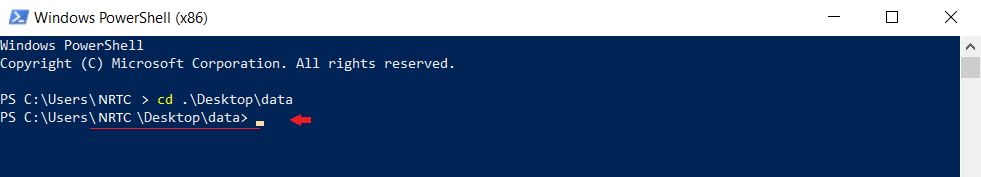
ودستورات زیر را وارد کنید:
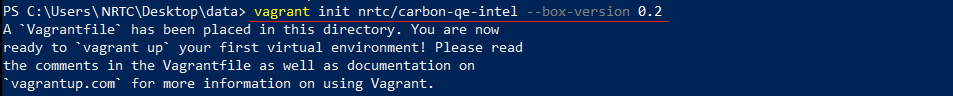
$ vagrant init nrtc/carbon-qe-intel --box-version 0.2
$ vagrant up
![]()
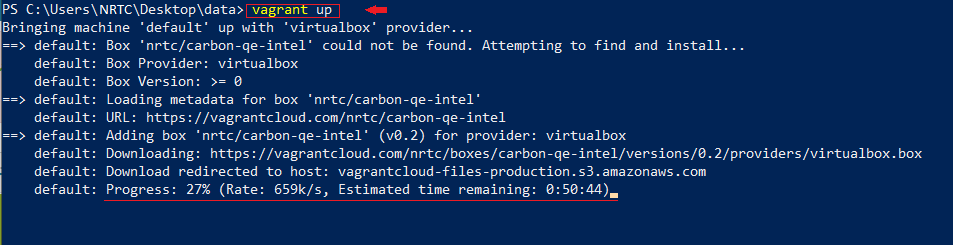
این مرحله نیازمند اندکی حوصله است تا باکس کربن از vagrant cloud روی سیستم شما بارگذاری شود.
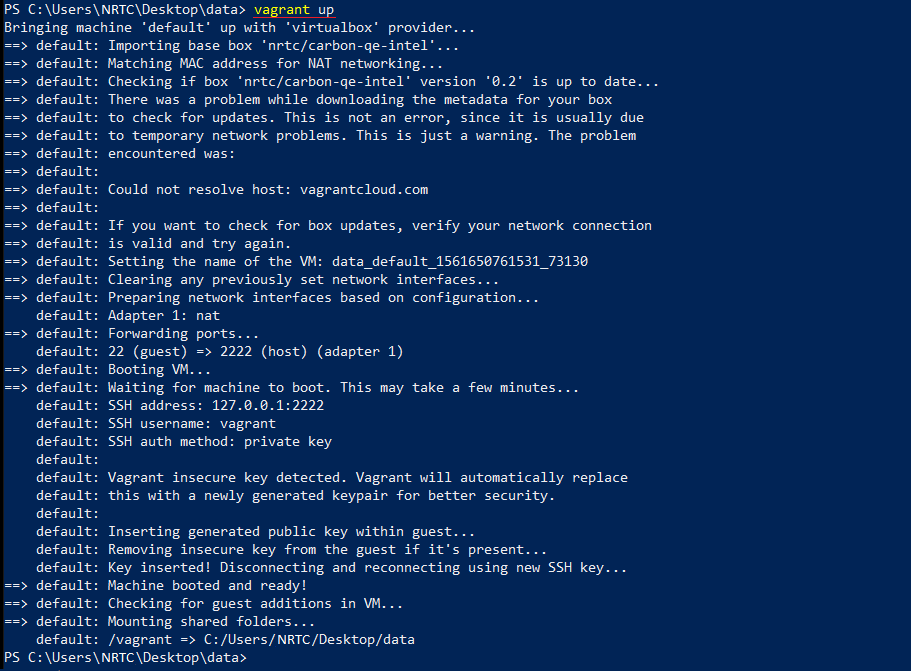
حال، توزیع محاسباتی CARBON بر روی رایانه شما راه اندازی شده است. فقط کافی است تا با دستور زیر وارد محیط CARBON شوید و مدیریت ماشین محاسباتی خود را در دست بگیرید:
$ vagrant ssh
![]()
میبینید که خطفرمان شما از محیط سیستم عامل اصلی، به محیط سیستمعامل محاسباتی CARBON تبدیل شده:
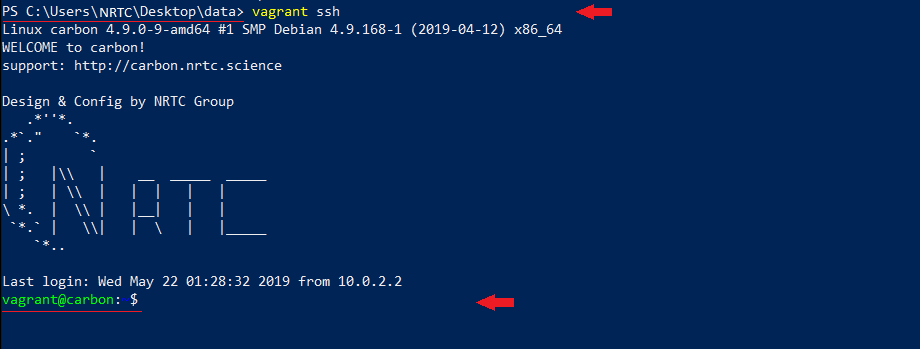
شما در محیط جدید، بدون نیاز به طی کردن مسیر پردردسر نصب برنامه، از نرم افزار محاسباتی مد نظر به سادگی استفاده میکنید.
برای اینکه به دادههای خروجی و نتایج محاسبات خود در سیستم عامل اصلی هم ببینید، باید تمام اطلاعاتتان را در ماشین مجازی، در دایرکتوری /vagrant ذخیره کنید تا در همان مسیر \data در سیستم عامل اصلی به آن دسترسی داشته باشید.
vagrant@nrtc/carbon-qe-intel:$ ~/vagrant

برای خروج از CARBON و دسترسی به خطفرمان سیستم اصلی خود،کافی است که exit را در ترمینال تایپ کنید:
$ exit

و یا از کلید میانبر "ctrl+D" استفاده کنید.
پس از خروج از ماشین مجازی CARBON، برای خاموش کردن ماشین، از دستور زیر استفاده کنید.
$ vagrant halt
![]()
دادههای محاسباتی و همه تغییرات شما در سیستم عامل مجازی CARBON ذخیره شده است.
برای ورود و خروجهای بعدی کافی است دستورات زیر را پیش بگیرید:
$ vagrant up
$ vagrant ssh
انجام محاسبات
$ exit
$ vagrant halt
نحوه درمیان گذاشتن مشکلات CARBON با ما
هرگونه مشکلی را که در حین کار کردن با CARBON به آن برخوردید، میتوانید از طریق github با ما در میان بگذراید.
برای اینکار باید در ریپازیتوریِ carbon-linux موجود در اکانت گیتهابِ NRTC ، یک issue ارسال کنید.
مراحل انجام اینکار را با هم دنبال می کنیم:
برای این کار وارد اکانت github خود شده و به صفحه اصلی NRTC به آدرس زیر بروید: https://github.com/NRTC
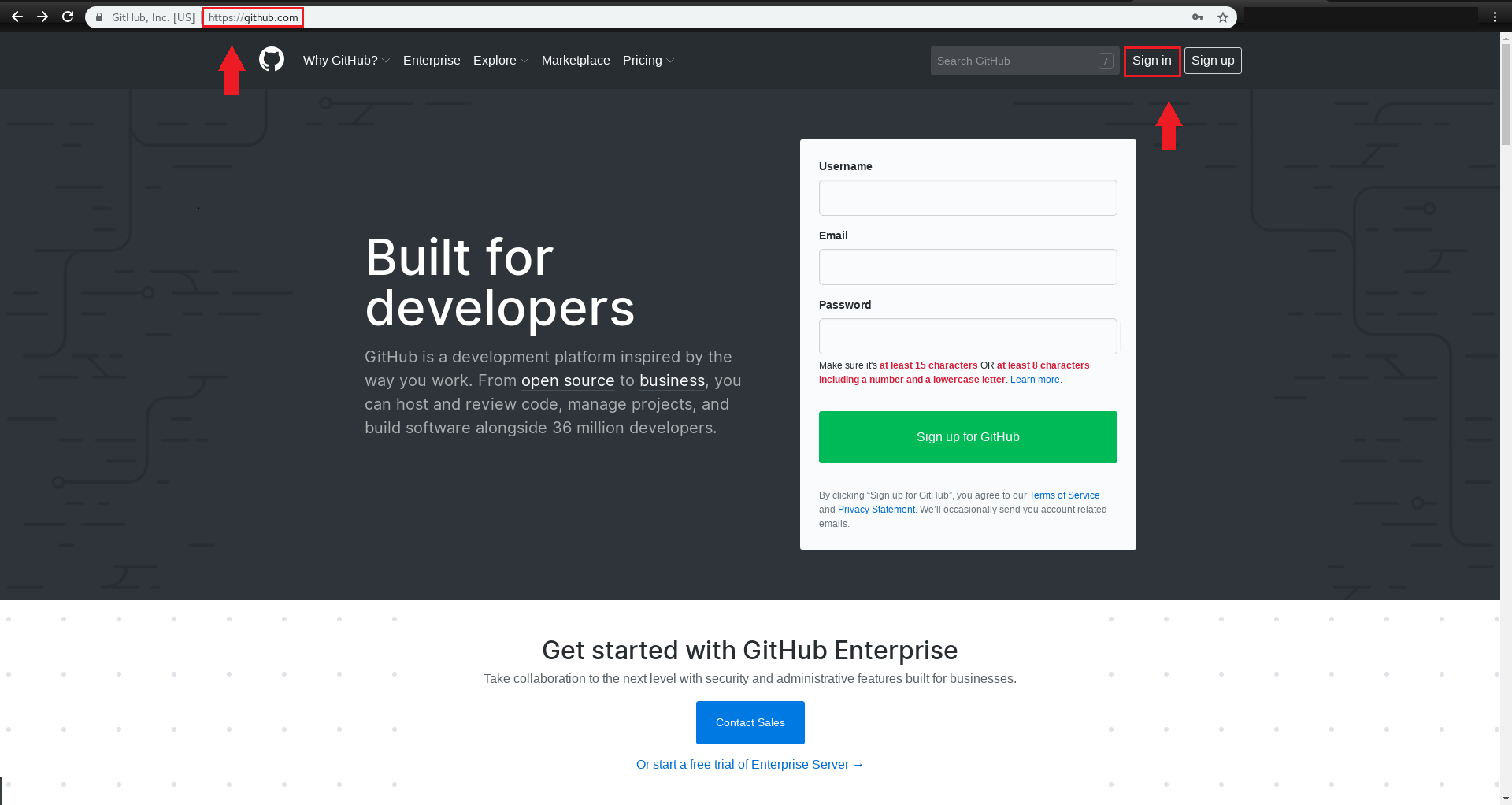
به ریپازیتوری carbon-linux وارد شوید
https://github.com/NRTC/carbon-linux
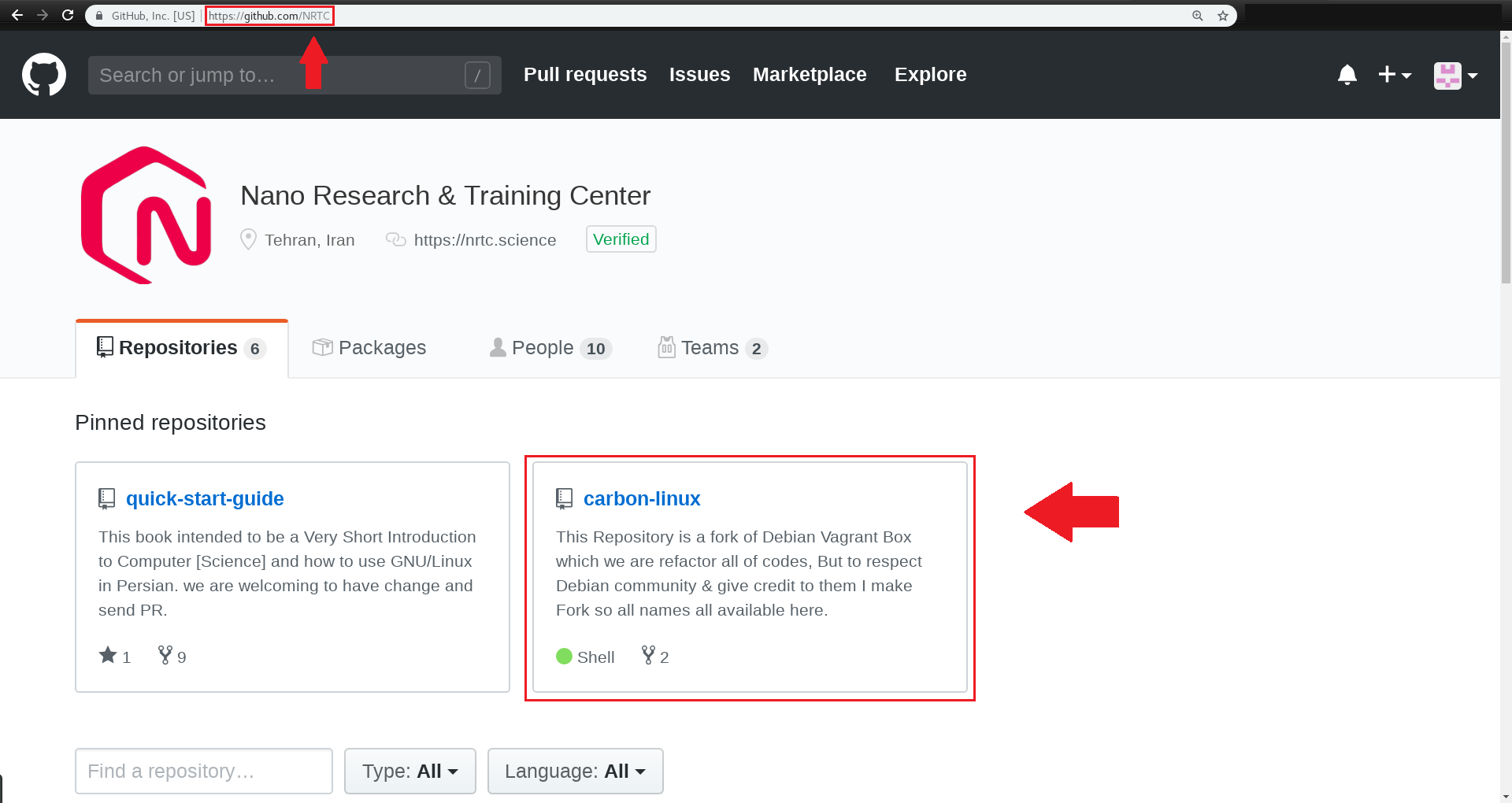
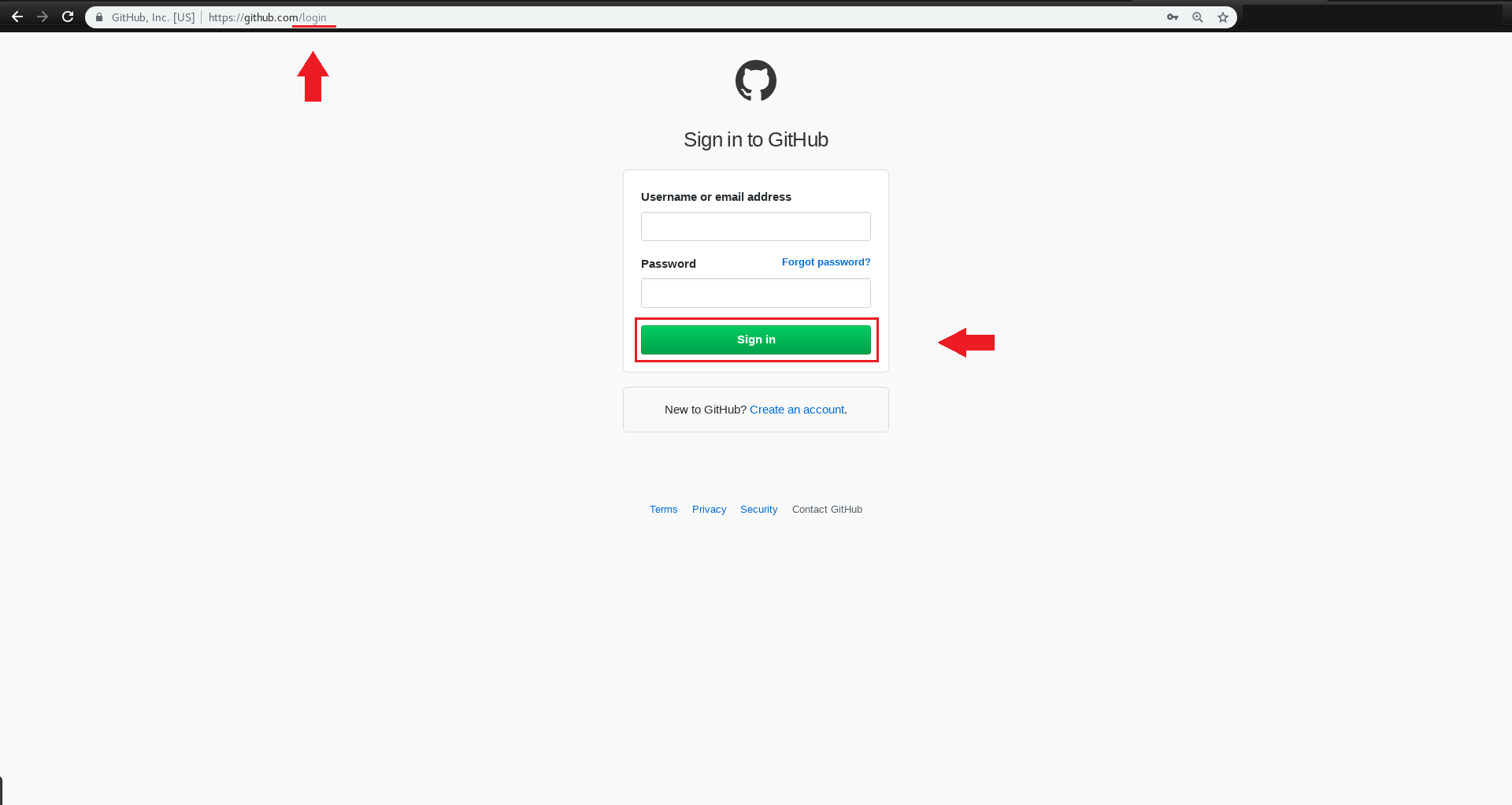
و پایین تر از اسم مخزن، روی issue کلیک کنید.
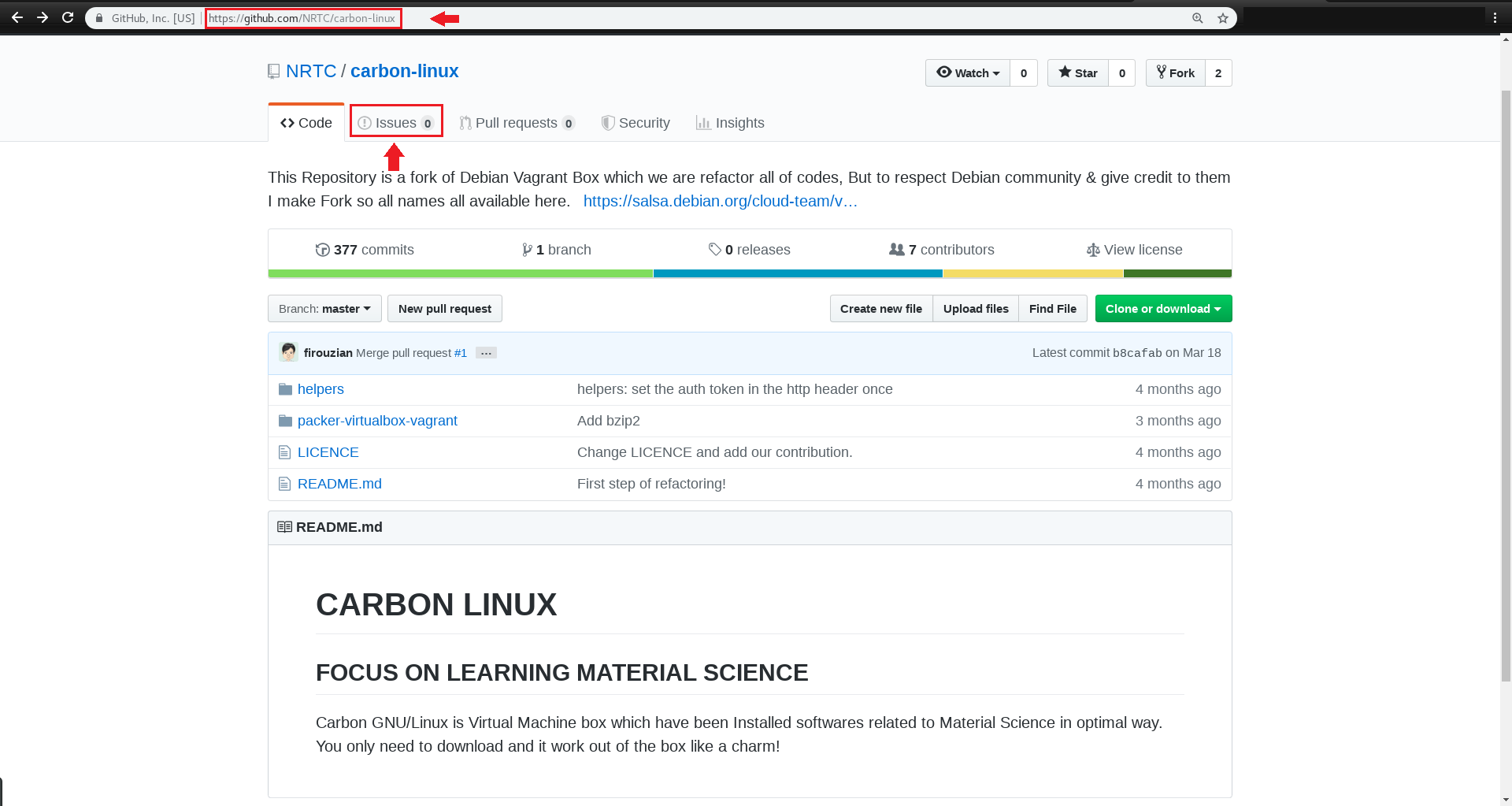
یک New Issue ایجاد کرده و عنوان و توضیح مشکل مدّنظر را بنویسید،
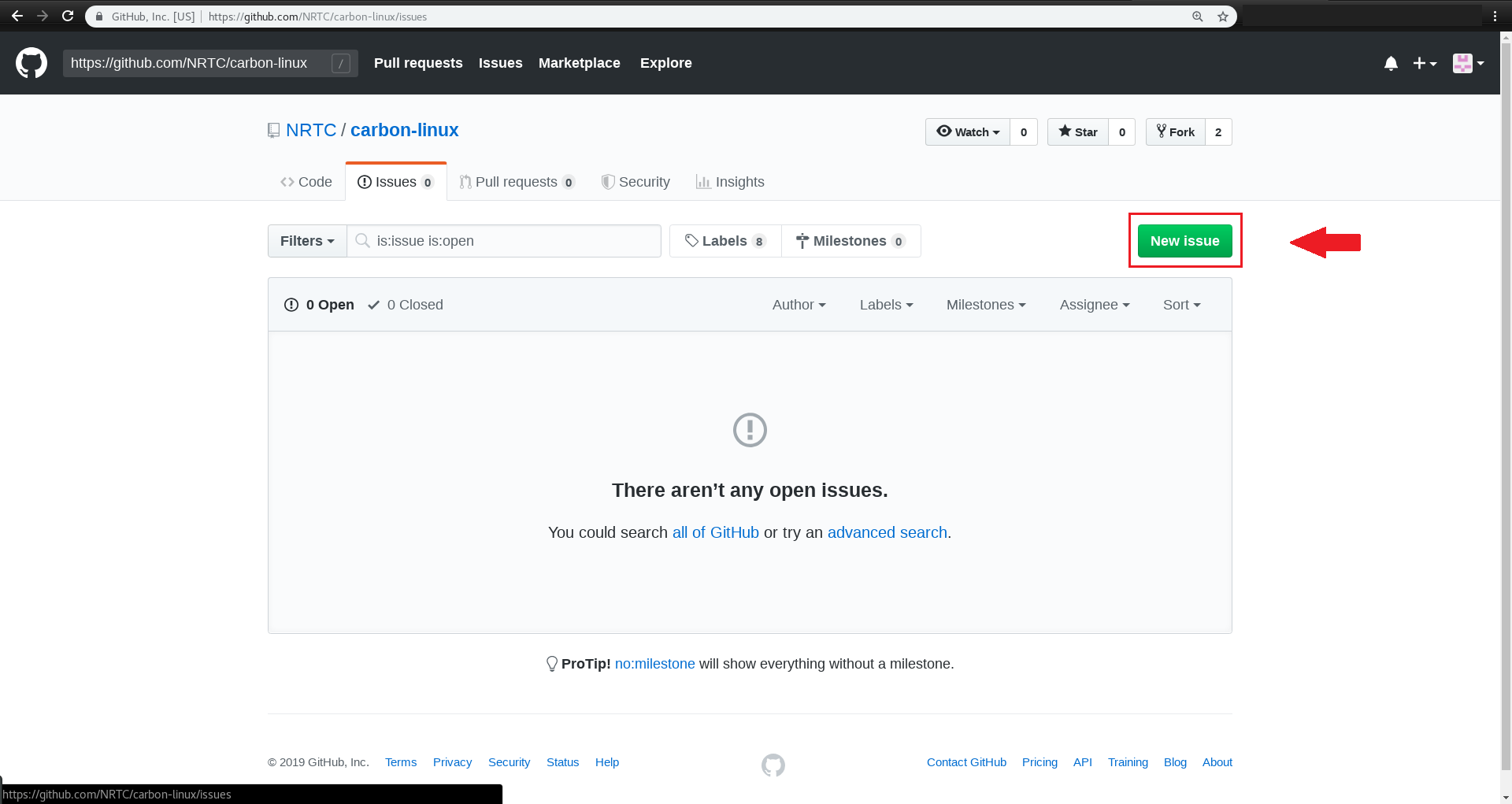
و در پایان روی Submit new issue کلیک کرده و آن را ارسال کنید.
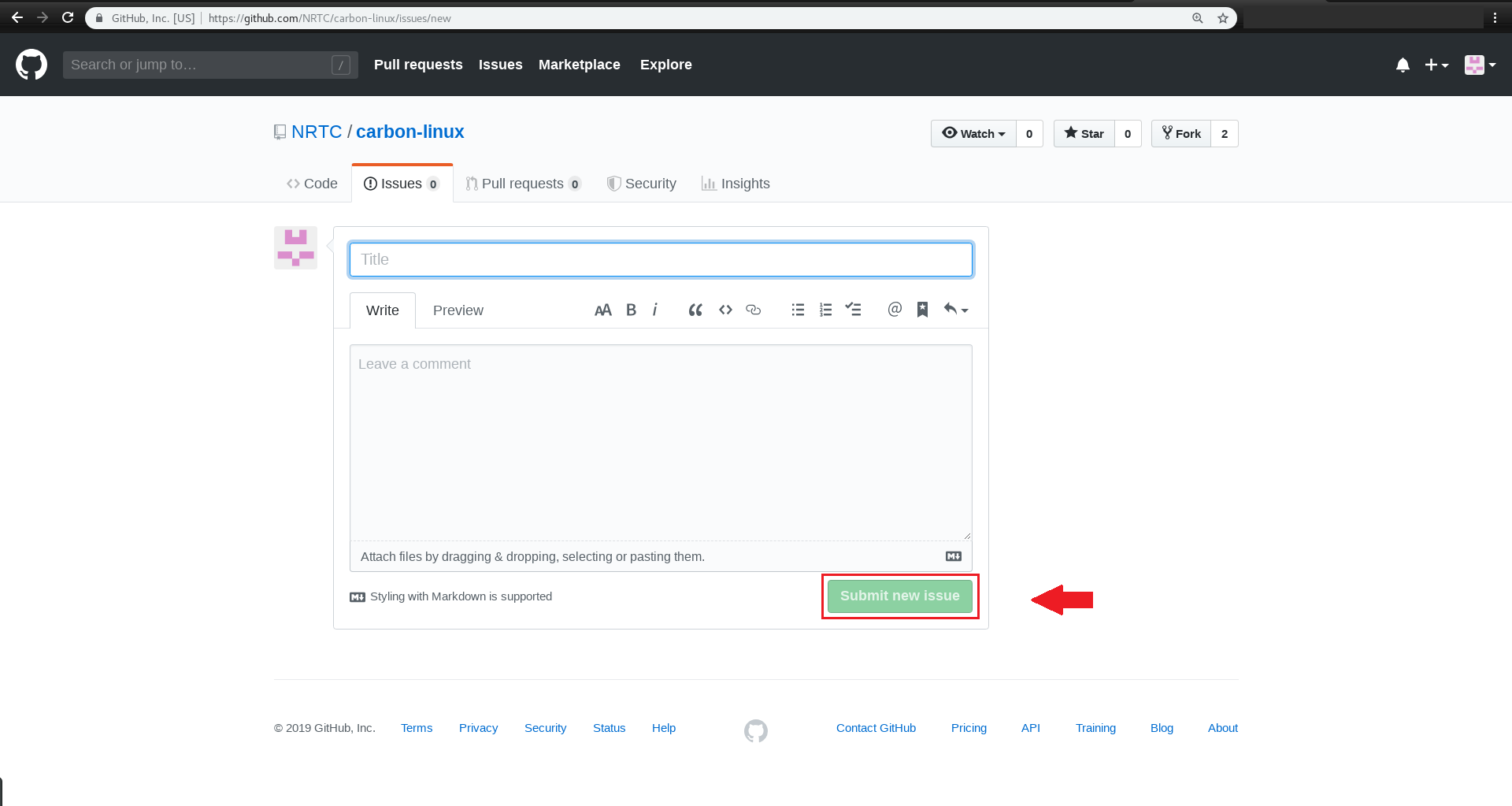
با این روش میتوانید، انتقاد و پیشنهادات و سایر درخواستهای خود را با ما مطرح کنید.


مرکز محاسباتی NRTC در نظر دارد با همکاری برخی از دانشگاه های معتبر کشور اقدام به برگزاری دوره های آموزشی نرم افزارهای شبیه سازی و محاسباتی نماید. علاقه مندانی که توانایی و تبحر کافی در زمینه ی نرمز افزارهای مختلف دارند می توانند بصورت رایگان از سامانه ی آموزشی این مرکز استفاده کرده و دوره های آموزشی مورد نظر خود را با این سامانه برگزار نمایند. این مرکز همچنین امکان صدور و ارائه ی گواهی معتبر و قابل ترجمه ی مدرس و گواهی دانشجو را دارا می باشد. علاقه مندان می توانند با مکاتبه با ایمیل زیر از جزئیات این طرح مطلع شده و هماهنگی های لازم را برای شروع هرچه سریعتر دوره های مورد نظر خود انجام دهند.
ایمیل مرکز محاسباتی NRTC:
This email address is being protected from spambots. You need JavaScript enabled to view it.


Current-voltage characteristics of armchair and zigzag γ-graphyne nanotubes with three different diameters under uniaxial strain are investigated by using first-principles quantum transport calculations through density functional theory (DFT) and non-equilibrium Green's function (NEGF) method. It is shown that for a given value of bias voltage, the resulting current depends strongly on the applied load so that tensile and compressive strain can generate Negative Differential Resistance (NDR) mostly into the armchair nanotubes. Our study reveals that the rectification behavior of the systems is mainly due to the asymmetric electron transmission function distribution in the conduction and valence bands.
Ref: https://iopscience.iop.org/article/10.1088/2053-1591/aafc59/meta


In this paper, the electronic and optical properties of single-layer (SL) and multilayer (ML) structures of MAPbX3 (X = Cl, Br, I and MA = CH3NH3) have been studied by density functional theory (DFT) in order to predict its photovoltaic capabilities. The results have shown that SL- and ML-MAPbX3 have a direct band gap in the range of 1.76–2.70 eV.
The calculation of dielectric constants has depicted that the static dielectric constants (SDCs) of SL-MAPbX3 are smaller than SDCs of ML-MAPbX3. However, as we expected, the reaction of the structures to in-plane (║) and out-of-plane (┴) polarizations was different; therefore, the SL- and ML-MAPbX3 (X = Cl, Br, I) were optically anisotropic.
In addition, the intensity of the optical absorption spectrum for ML-MAPbX3 structures is approximately three times higher than that of SL-MAPbX3 structures. By increasing the radius of halogens (RCl<RBr<RI), surface area under the absorption curve increases and absorbs more.
Furthermore, our results have shown that the electronic and optical behavior of 2D-MAPbX3 is suitable for photovoltaic applications and makes them useful for OIHP solar cells.
Ref: https://www.tandfonline.com/doi/abs/10.1080/15567036.2019.1568645


In this study, using the density functional theory, the mechanical properties of methylammonium lead halide perovskites (CH3NH3PbX3, X = I, Br, Cl) were investigated. Young’s modulus, bulk modulus, and shear modulus, Poisson’s ratio, and many other parameters were calculated using the PBEsol and vdW approximations. Also, in this work, utilizing a new accuracy in calculating the elastic constants, the intense conflict between the previous theoretical results and the experimental data were fixed. Moreover, for the first time, through combination of the PBEsol and vdW methods, the effect of the interaction between methylammonium and PbX3 scaffold on the mechanical properties of lead halide perovskites was well cleared. In continuation, using the PBEsol+vdW method, a phase transition appeared for the MAPbBr3 and MAPbCl3 structures, which proved more stability of MAPbBr3 and MAPbCl3 in comparison with MAPbI3. In what follows, by studying these materials under an applied strain beyond the harmonic region, the transition zone to the plastic area in the strain region of 5.5% and smaller was identified, and the small values of the aforementioned applied strains were found to be the reason for the instability of these materials at room temperature and above.
Ref: https://pubs.acs.org/doi/abs/10.1021/acs.jpcc.7b07129


Ref: https://ieeexplore.ieee.org/abstract/document/8617679


By means of first-principles calculations, the mechanical properties and the strain-dependent electronic band structure of the orthorhombic SnS monolayer were investigated. In an attempt to investigate the elasticity of this material, six deformation modes were considered. The stability of this configuration against these external tensions was assessed, and the second-order elastic constants were found to be C11 = 45.2 N/m, C22 = 25.3 N/m, C12 = 18.0 N/m, and C66 = 54.3 N/m. The third- and fourth-order elastic constants, which shed light on the material behavior at strains above the harmonic region, were also determined. The stress-strain relationships imply that the SnS monolayer can withstand tensile strains up to 0.075, 0.29, and 0.12 for uniaxial strain along the a direction, uniaxial strain along the b direction, and biaxial strain, respectively. Moreover, a study of the electronic band structure of the SnS monolayer shows that this indirect band gap semiconductor when exposed to a marginal uniaxial or biaxial strain (about 2%) experiences a transition to a direct band gap semiconductor with a moderate band gap (<2.7 eV). This transition merely occurs when tensile strain is exerted. Our findings suggest that under compressive strain, this semiconducting material maintains its indirect band gap nature, and the band gap predominately declines. Lastly, the band splitting that arises from spin-orbit interactions in most cases vanishes when strain is applied. However, compressive strain along the a direction and tensile strain along the b direction are two exceptions, in which the former strengthens the spin-orbit effects and for the latter, band splitting remains almost unchanged.
Ref: https://www.sciencedirect.com/science/article/abs/pii/S0022369718302075


In this paper, we have addressed the structural stability and hydrogen storage capability of single side and double side Na-decorated γ-Graphyne, Graphyne-like Boron Nitride and CCBN-yne, using three methods of density functional theory approximation: PBEsol, vdW-DF2-B86R and vdW-DF2. We also investigated the electronic properties of these structures, decorated by Na atoms and adsorbing hydrogen, such as density of states and band structure. Our results showed that hydrogen can be adsorbed up to three molecules per Na atom. We found the optimal geometries of adsorbed hydrogen molecules on adsorption candidates. We also found that Direct band-gap at “M” point changes after decoration and adsorption in mentioned structures. Finally, we explored metallic structures for hydrogen storage which can be used in the industry of fuel cells.
Ref: https://www.sciencedirect.com/science/article/pii/S0025540817310139


In this paper, the mechanical properties of Na and Pt decorated arrays of 2D graphyne sheet is investigated. The proposed structures are consisted of Na and Pt decorated graphyne sheet (CC), analogous system of Boron nitride sheet (BN-yne), and graphyne-like BN sheet (CC-BN-yne). The properties such as In-plane stiffness and Bulk module are studied using Energy-Strain correlation. The calculations were carried out based on Density functional theory (DFT) using the generalized gradient approximation (GGA) framework. The results offered very competitive values of stiffness and Bulk module for Pt decorated CC and BN-yne. However, the Pt decorated CC-BN-yne structure demonstrated around 80% of stiffness and 77% of Bulk module, compared to those of pure structure. Na decorated system showed the same trend for all three mentioned structures.
Ref: https://www.sciencedirect.com/science/article/pii/S0749603616309028
![]()






